- О компании
- Направления деятельности
-
Медиатека
СтатьиСервис

10.06.2025
Сервисная сторона лаборатории: от инсталляции оборудования до постгарантийной поддержки
Интервью со специалистами технической поддержки Компании Хеликон.
СтатьиИнтервью
27.03.2024
Интервью Анатолия Смирнова телеканалу PROБизнес
«В любой биологической лаборатории в России есть что-то от нас».
- Новости
- Мероприятия
- Партнеры




.jpg "Реализованные проекты")
Медиатека
stLFR – технология секвенирования длинных фрагментов ДНК от MGI на базе платформ второго поколения
Оглавление:
•
Три поколения методов секвенирования ДНК
•
О технологии stLFR
•
Подготовка библиотек
Этап 1. Вставка транспозона
Этап 2. Гибридизация ДНК с микрочастицами
Этап 3. Лигирование баркода
Этап 4. Расщепление избыточных олигонуклеотидов, лигирование второго адаптера, ПЦР
•
Возможности использования
1. Анализ небольших генетических вариаций
2. Фазирование гаплотипов
3. Обнаружение структурных вариаций в геноме
4. Ресеквенирование и de novo сборка геномов
•
Примеры использования
Этап 2. Гибридизация ДНК с микрочастицами
Этап 3. Лигирование баркода
Этап 4. Расщепление избыточных олигонуклеотидов, лигирование второго адаптера, ПЦР
2. Фазирование гаплотипов
3. Обнаружение структурных вариаций в геноме
4. Ресеквенирование и de novo сборка геномов
Три поколения методов секвенирования ДНК
За более чем 40 лет существования технологии секвенирования претерпели существенную эволюцию, в ходе которой кардинально менялись принципы их работы, стоимость, время- и трудозатратность, а также производительность. Сейчас выделяют три основных поколения секвенирования, которые характеризуются разными показателями и используются для разных целей.
К первому поколению относят методы, которые в свое время стали революционными и позволили впервые расшифровать последовательности ДНК. Это самый известный метод секвенирования по Сэнгеру (метод обрыва цепи), а также менее распространенный метод химической деградации Максама-Гилберта. Метод Сэнгера со временем значительно оптимизировали и автоматизировали, но он всё ещё остается довольно дорогим, времязатратным и низкопроизводительным, хотя и очень точным, а предел длины фрагментов ДНК для секвенирования составляет 1000 пар оснований (п.о.). Сейчас платформы на основе метода Сэнгера применяют в основном для рутинного секвенирования коротких фрагментов ДНК и валидации данных, полученных другими методами.

Технологии второго поколения, более известные под аббревиатурой NGS (next generation sequencing, секвенирование следующего поколения) отличались более простым процессом секвенирования. Это привело к научному прорыву в геномике и смежных областях. Одно из их главных преимуществ – высокая производительность, позволяющая одновременно прочитывать миллионы и даже миллиарды последовательностей ДНК, длина которых в зависимости от метода варьируется от 30 до 500 п.о. Платформы секвенирования второго поколения основываются на разных принципах и подходах: пиросеквенирование, секвенирование путем синтеза с обратимой терминациией, ионное полупроводниковое секвенирование, лигазное секвенирование и другие, но большинство из них объединяет этап пробоподготовки, включающий стадию амплификации фрагментов (ПЦР).
Революционность методов третьего поколения, которые также относят к NGS, состоит в том, что они позволяют напрямую секвенировать длинные единичные молекулы ДНК (длина получаемых прочтений достигает от 1 тыс. до 2 млн. п.о.), избегая стадии амплификации. Это значительно упрощает ресеквенирование геномов или их сборку de novo, а также позволяет преодолеть сложности сборки данных, полученных методами второго поколения (например, невозможность разделять повторяющиеся последовательности или крупные геномные перестройки). К технологиям третьего поколения относят одномолекулярное секвенирование в реальном времени (SMRT), нанопоровое секвенирование, ко-баркодирование и другие 1, 2.

Одной из базовых характеристик методов секвенирования является длина фрагментов. В результате их секвенирования получают нуклеотидные последовательности, или прочтения (риды). Методы второго поколения относятся к технологиям получения коротких прочтений (от 30 до 500 п.о.), третьего поколения – длинных прочтений (от 1 тыс. до 2 млн п.о.). Современные гибридные подходы сочетают преимущества технологий сразу двух поколений секвенирования.
Компания MGI предлагает технологию stLFR, которая позволяет получать длинные высокоточные прочтения, сочетая экономичность и эффективность технологий секвенирования второго поколения.
О технологии stLFR
В основе технологии stLFR (single-tube long fragment read, чтение длинных фрагментов в одной пробирке) лежит принцип ко-баркодирования ДНК – добавления одного и того же баркода к субфрагментам длинных одиночных молекул ДНК. Уникальные баркоды (от 30 до 50 млн в одном образце) расположены на поверхности микрочастиц и позволяют кодировать миллионы коротких фрагментов ДНК, которые затем анализируются на платформах секвенирования второго поколения. После секвенирования исходные длинные молекулы ДНК реконструируются на основе полученных прочтений и баркодов.
Преимущества stLFR:
- получение длинных прочтений (средняя длина – 50-60 тыс. п.о., максимальная – до 300 тыс. п.о.) на платформах секвенирования второго поколения;
- простота исполнения и экономичность;
- замена стандартных методов подготовки библиотек для секвенирования второго поколения;
- реализация на стандартном оборудовании вне зависимости от технологии секвенирования;
- весь рабочий процесс в одной пробирке;
- требование небольшого количества ДНК (1-10 нг) 3.
Подготовка библиотек
Этап 1. Вставка транспозона
Первым шагом в stLFR является вставка транспозазой Tn5 транспозона с гибридизационной последовательностью вдоль длинных молекул ДНК (геномной ДНК) примерно каждые 200-1000 п.о. (Рис. 1). Эта последовательность будет использоваться для связывания длинного фрагмента ДНК с баркодом. Транспозаза остается связанной с длинной молекулой ДНК, поддерживая её структуру.
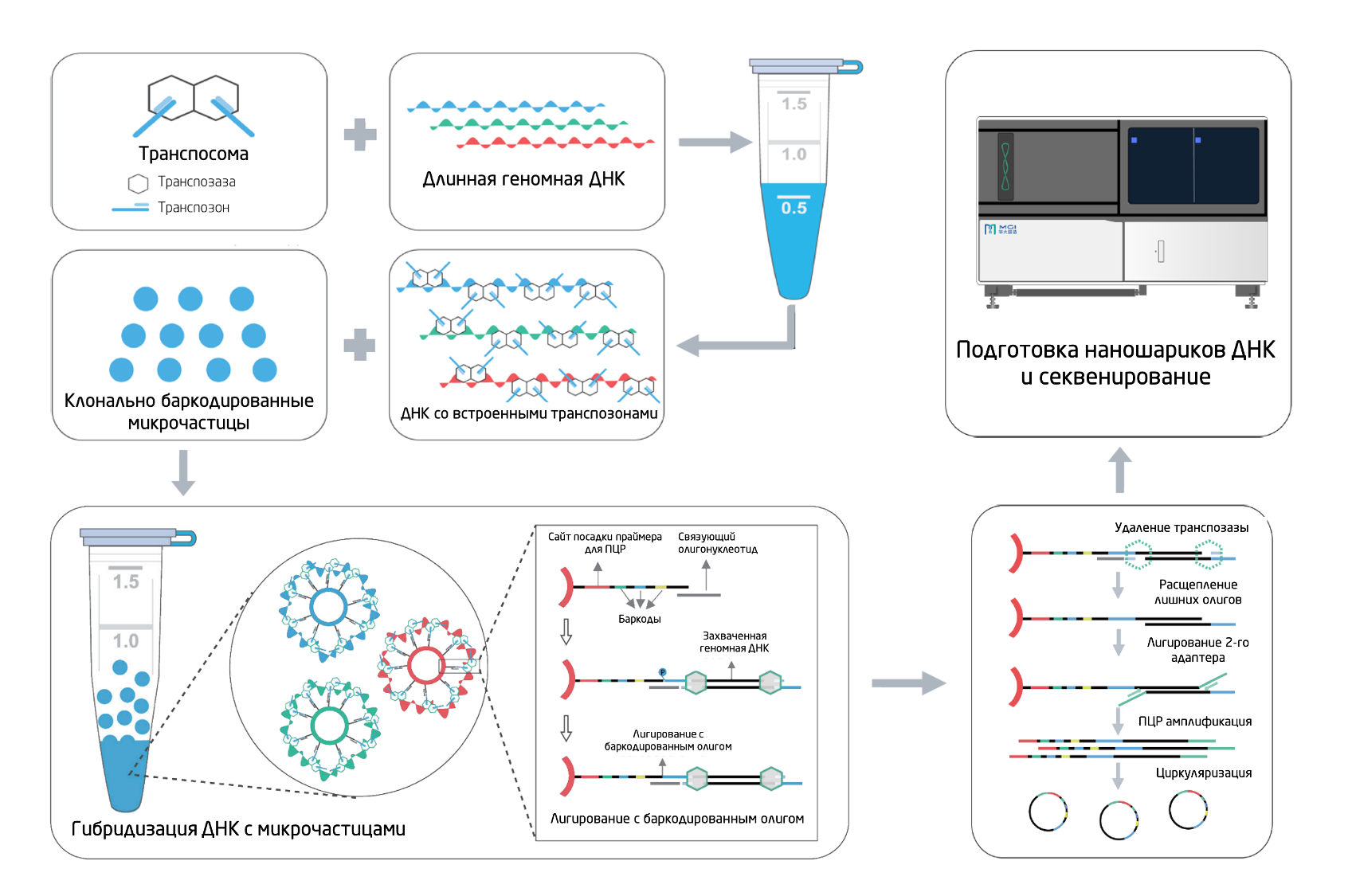
Рис. 1. Схематическое представление метода stLFR.
1) Вставка транспозона, содержащего гибридизационную последовательность для последующего связывания с баркодом, в длинные молекулы ДНК.
2) Гибридизация молекул ДНК с микрочастицами, покрытыми уникальными олигонуклеотидами, которые содержат уникальный баркод, сайт для связывания праймеров для ПЦР и связующий олигонуклеотид, комплементарный гибридизационной последовательности транспозона.
3) Лигирование транспозонов с баркодированными олигонуклеотидами.
4) Расщепление избыточных олигонуклеотидов, лигирование второго адаптера, ПЦР. Секвенирование готовой библиотеки осуществляется на секвенаторе DNBSEQ-G400, MGI.
Этап 2. Гибридизация ДНК с микрочастицами
На втором этапе к ДНК добавляют микрочастицы с уникальными баркодированными олигонуклеотидами (30-50 млн микрочастиц в образце с 400.000 копий на каждую) и происходит связывание гибридизационной последовательности транспозона с комплементарной областью связующего олигонуклеотида. За счет изменения вторичной структуры молекула ДНК наматывается на микрочастицу, обеспечивая равномерное и высокоэффективное связывание.
Этап 3. Лигирование баркода
Следующим шагом отдельные последовательности баркодов переносятся на каждый субфрагмент ДНК посредством лигирования разрыва между гибридизационной последовательностью и связующим олигонуклеотидом. В этот момент комплексы ДНК/транспозазы разрушаются, образуя субфрагменты размером меньше 1 тыс. п.о.
Этап 4. Расщепление избыточных олигонуклеотидов, лигирование второго адаптера, ПЦР
Из-за большого числа микрочастиц и высокой плотности расположения количество олигонуклеотидов на несколько порядков превышает количество продукта. Чтобы не перегружать следующие шаги протокола, неиспользованные избыточные олигонуклеотиды расщепляются за счет специфической экзонуклеазной активности.
Для достижения максимального покрытия последовательностей ДНК используется подход с добавлением второго адаптера путем неканонического лигирования. Теоретически это позволяет амплифицировать и секвенировать все субфрагменты гибридизованной молекулы ДНК. Кроме того, этот этап позволяет разместить баркод образца рядом с геномной последовательностью для мультиплексирования образца. Это может быть полезно, т.к. для считывания этого баркода не потребуется дополнительный праймер для секвенирования. После этапа лигирования второго адаптера выполняется ПЦР.
Полученная библиотека готова к секвенированию на секвенаторе DNBSEQ-G400, MGI 3.
Возможности использования
Методология stLFR активно внедряется в исследовательскую практику и может использоваться для следующих направлений исследований:
1. Анализ небольших генетических вариаций
Наравне с технологиями получения коротких прочтений stLFR позволяет обнаруживать небольшие изменения геномных последовательностей, таких как единичные нуклеотидные полиморфизмы (SNPs) и индели.
2. Фазирование гаплотипов
При секвенировании диплоидных геномов могут быть получены прочтения, соответствующие двум наборам родительских хромосом. Проблема фазирования, т.е. определения принадлежности аллеля одной из пары хромосом, может быть решена с помощью stLFR.
3. Обнаружение структурных вариаций в геноме
stLFR позволяет идентифицировать более протяженные геномные перестройки, такие как инверсии, делеции, траснлокации, инсерции, недоступные для обнаружения с помощью технологий секвенирования второго поколения.
4. Ресеквенирование и de novo сборка геномов
stLFR показал свою эффективность в секвенировании полных геномов.
Примеры использования
С помощью stLFR впервые был de novo секвенирован геном рыбы-ежа 4. Эти рыбы известны своим интересным защитным механизмом – в момент опасности они наполняют желудок водой и раздуваются до шарообразной формы, выставляя наружу острые иглы. Прочтение их генома имеет важное значение для дальнейших исследований онтогенеза, филогенеза и эволюции этих необычных морских обитателей.
В другой работе 5 внимание было обращено к еще одной группе удивительных созданий – бактериям-экстремофилам. С применением stLFR и других технологий ученые отсеквенировали и собрали геномы пяти штаммов устойчивых к радиации экстремофилов. Рабочий процесс stLFR для получения бактериальных геномов высокого качества в масштабе хромосомы был оптимизирован от стадии подготовки библиотек до сборки de novo. В дальнейшем этот рабочий процесс можно будет использовать для быстрого и точного секвенирования любых целевых штаммов, в том числе сложно культивируемых и дающих маленький выход ДНК из-за сложностей экстракции.

Технология stLFR показала отличные результаты в сравнении с другими методами получения длинных прочтений при секвенировании полного растительного генома, редкого подвида ореха макадамия 6. Он оказался самым дешевым и генерировал сборку с наименьшим количеством единичных ошибок и инделей.

Литература
1. Dorado G. et al. Analyzing Modern Biomolecules: The Revolution of Nucleic-Acid Sequencing – Review. Biomolecules, 2021.
2. Verma M., Kulshrestha S., Puri A. Genome Sequencing. Methods in Molecular Biology, 2017.
3. Wang O. et al. Efficient and unique cobarcoding of second-generation sequencing reads from long DNA molecules enabling cost-effective and accurate sequencing, haplotyping, and de novo assembly. Genome Research, 2019.
4. Xu M.Y. et al. Draft genome of a porcupinefish, Diodon Holocanthus. bioRxiv, 2019.
5. Zhang Z, et al. Comparison of different sequencing strategies for assembling chromosome-level genomes of extremophiles with variable GC content. iScience, 2021.
6. Murigneux V. et al. Comparison of long-read methods for sequencing and assembly of a plant genome. Gigascience, 2020.