.jpg "Реализованные проекты")


Медиатека
Определение пространственных структур белков методом ЯМР спектроскопии
Статья Павла Миронова, студента магистерской программы «Структурная биология и биотехнология» МГУ имени М. В. Ломоносова и стипендиата Компании Хеликон.
Компания Хеликон всегда стремилась поддерживать отечественных учёных – как опытных специалистов, так и студентов, которые только делают первые шаги в науку. С этой целью мы учредили ежегодную стипендию «Хеликон» для молодых биологов со всей страны. Первыми стипендиатами стали студенты магистерской программы «Структурная биология и биотехнология», реализуемой на базе МГУ имени М.В. Ломоносова.
Предлагаем вам прочесть статью Павла Миронова об истории и современном использовании одного из самых передовых методов исследования структур биологических макромолекул — спектроскопии ядерного магнитного резонанса.
Во второй половине XX века единственным методом получения пространственных структур белков с атомным разрешением являлся рентгеноструктурный анализ (РСА). Первая рентгеновская структура белка была получена Максом Перуцем в 1957 году для миоглобина. 1 В это же время в области структурной биологии активно развивался метод спектроскопии ядерного магнитного резонанса (ЯМР), который был пригоден лишь для изучения низкомолекулярных соединений из-за своей низкой чувствительности. Начиная с 70-х годов, в области ЯМР стали применяться методы Фурье-преобразования, появились стабильные магниты с сильным полем, что значительно повысило чувствительность и разрешение ЯМР, позволив обрабатывать многомерные спектры крупных молекул. Наконец, в 1985 году химику Курту Вютриху и его коллегам удалось впервые получить пространственную структуру белка ингибитора протеиназы IIА в водной среде методом ЯМР на основе разработанного вычислительного подхода 2, за что он был удостоен Нобелевской премии по химии в 2002 году. Достижение Вютриха показало возможность изучения структуры белков в водной среде в наиболее естественных физико-химических условиях, в отличие от кристаллической формы, требуемой в РСА. С этого момента ЯМР стал мощным фундаментальным методом для определения трехмерных структур биологических макромолекул.
Как из пробирки с водным раствором белка получить его 3D модель?
Первым этапом в расчете структуры белка является работа с ЯМР-спектрами. Для немеченых белков обычно используют двумерные ЯМР-спектры TOCSY (TOtal Correlated SpectroscopY) и NOESY (Nuclear Overhauser Effect SpectroscopY) (Рис. 1A, 2A). В них записываются сигналы, которые показывают взаимодействие двух ядер ¹Н в молекуле между собой. В спектрах TOCSY сигнал формируется за счет спин-спинового взаимодействия, который основывается на переносе намагниченности между ядрами атомов, связанных ковалентными связями (Рис. 1АВ; пунктирные линии в 2В). В спектрах NOESY наблюдается диполь-дипольное взаимодействие, которое заключается в переносе намагниченности через пространство между спинами (Рис. 2АВ, пунктирные стрелки) из-за ядерного эффекта Оверхаузера (ЯЭО, NOE).
Процесс отнесения сигналов в спектре для начинающего специалиста по ЯМР кажется расшифровкой древней письменности племен майя. Для каждого сигнала в спектре вручную определяется его соответствие реальному атому в молекуле. Для начала в TOCSY отмечают все сигналы атомов водорода, взаимодействующих через ковалентные связи. Сигналы с ядра ¹HN на атомы водорода боковой цепи (обозначения HN/Hβ на Рис.1) позволяют выделить спиновые системы, которые соответствуют одному аминокислотному остатку (a.о.). (Рис. 1А). По химическим сдвигам сигналов внутри спиновой системы можно установить тип остатка.
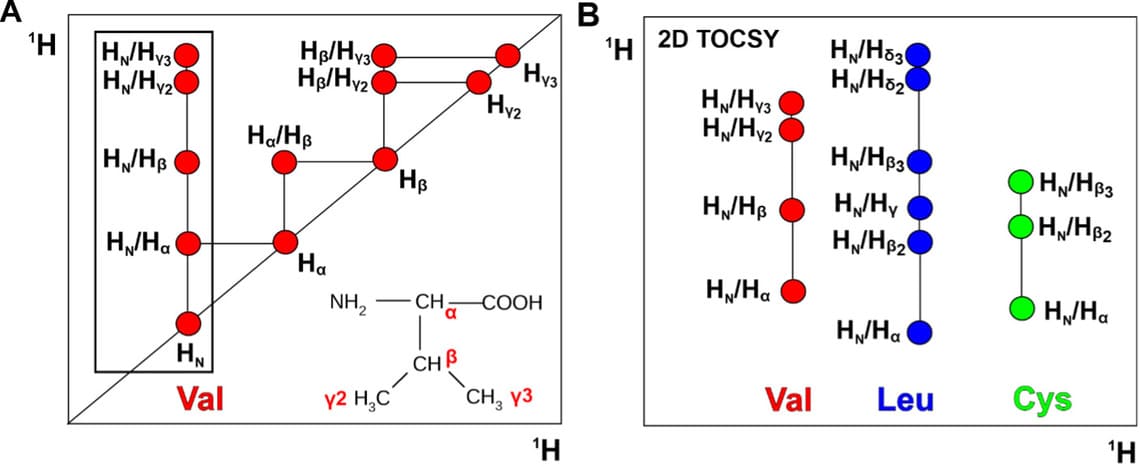
Рисунок 1. A. Схематическое изображение 2D TOCSY спектра, показывающее расположение сигналов атомов валинового остатка (Val). Спиновая система выделена рамкой. B. Схема расположения сигналов спиновых систем в спектре 2D TOCSY. Показана область спектра для трех остатков гипотетического пептида. Резонансные сигналы атомов водорода валина, лейцина и цистеина обозначены красными, синими и зелеными кружками соответственно.
Чтобы соединить спиновые системы между собой в соответствии с сиквенсом белка, используют спектр NOESY, который показывает контакты через пространство между протонами на расстоянии меньше 6 ангстрем (Рис. 2А). Контакты между амидным протоном (HNⁱ) и атомами водорода предыдущего остатка (к примеру Hαⁱ-1) позволяют установить порядок спиновых систем, а также контакты между боковыми цепями соседних остатков (Рис. 2АB).
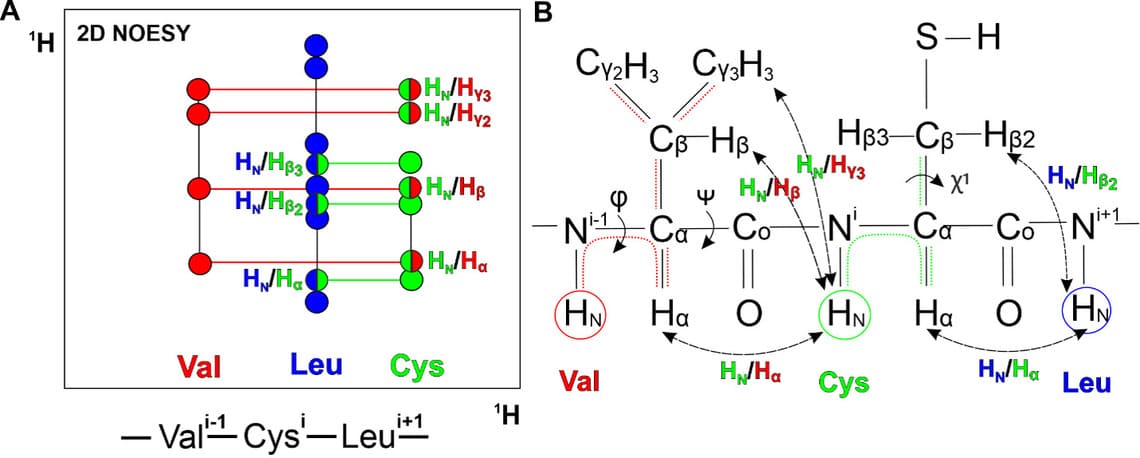
Рисунок 2. A. Область 2D NOESY спектра как на рис. 1В. Контакты между спиновыми системами отображены цветами двух взаимодействующих систем. B. Структурная формула сегмента валин-цистеин полипептидной цепи. Пунктирные линии соединяют группы атомов водорода, между которыми есть спин-спиновое взаимодействие не более чем через три химические связи. Пунктирные стрелки связывают пары атомов водорода в соседних аминокислотных остатках, для которых наблюдается NOE-эффект. Сплошные стрелки указывают на торсионные углы.
Следующим этапом является получение набора ограничений из обработанных спектров. Спектр NOESY позволяет получить ограничения на расстояния между взаимодействующими атомами в пространстве. Интенсивность сигналов в спектре пропорциональна межъядерному расстоянию как 1/R⁶. Чем интенсивнее сигнал между взаимодействующими ядрами, тем ближе расположены они друг к другу в пространстве. Ограничения на межпротонные расстояния являются основным параметром при расчете 3D-структуры белка. Спектр TOCSY дает информацию об ограничениях на торсионные углы, которые получают из констант спин-спиновых взаимодействий (КССВ) между амидным протоном HN и Hα для φ углов, а для χ¹ из КССВ между Hα и Hβ в спектрах для каждого а.о. 3 (Рис. 2В). Дополнительно можно вводить ограничения на водородные и дисульфидные связи.
Ограничения накладываются на модель молекулы в программе для расчета структуры. На основе вычислительного алгоритма ограничения на расстояния преобразуются в матрицу координат, по которым генерируются случайные конформации молекулы. 4 После происходит расчет МД-траектории молекулы с понижающейся температурой (отжиг). При понижении температуры системы происходит поиск оптимальной конформации белка с минимальной энергией. Ограничения выступают в роли потенциальной энергии, которая заставляет молекулу принять структуру, совместимую с данными ЯМР. В результате расчета получается несколько вариантов структур белка с оптимальной конформацией, удовлетворяющих экспериментальным ограничениям 5 (Рис. 3АВ).
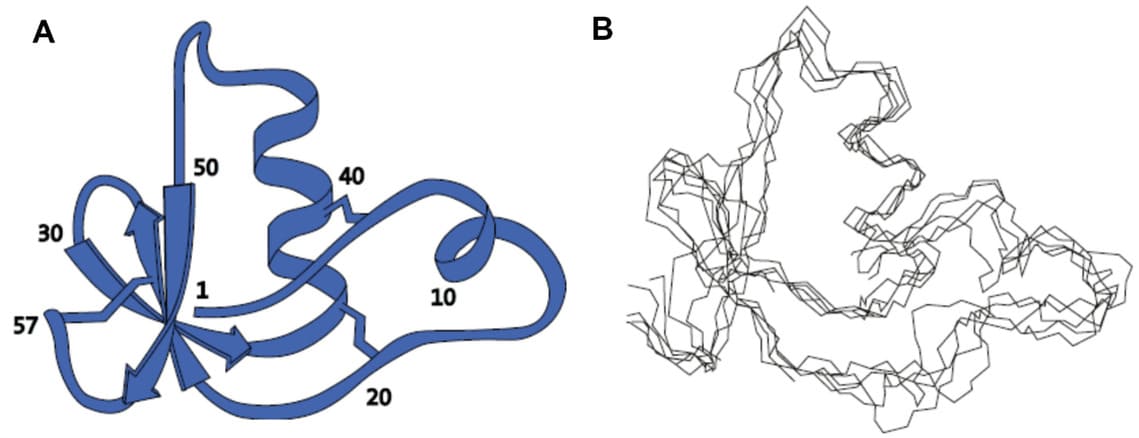
Рисунок 3. A. 3D структура ингибитора протеиназы IIА (BUSI IIA). Цифрами указаны номера остатков в полипептидной цепи. B. Набор пространственных структур, полученных в результате применения вычислительного алгоритма в программе DISGEO
2. Отображены только атомы основной цепи. Рисунок заимствован из
Advanced information. NobelPrize.org, 2002.
Используя вышеописанную методику, Вютрих получил первую в мире ЯМР-структуру белка ингибитора протеиназы IIА (BUSI IIA) 2 и показал, что структура BUSI IIA имеет сходную структуру с известными структурами гомологичных ему белов, полученных РСА. Чтобы доказать, что полученные в ходе расчета трехмерные структуры способны в точности характеризовать строение реальных молекул, для проверки достоверности была проделана та же вычислительная процедура для уже известной кристаллической структуры панкреатического ингибитора трипсина (PSTI). Из данных РСА сгенерировали ЯМР-ограничения для PSTI и рассчитали для него ЯМР-структуру, которая также показала хорошую сходимость с его кристаллической структурой. 6
В настоящий момент ЯМР-спектроскопия высокого разрешения является передовым методом в исследовании структуры, динамики и молекулярных взаимодействий биологических макромолекул в водном растворе. На данный момент в базе данных PDB насчитывается более 12 тысяч ЯМР-структур белковых соединений, и это число из года в год продолжает возрастать.
Автор: Миронов Павел
Литература
1. Kendrew, J. C. et al. A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis. Nature 181, 662–666 (1958).
2. Williamson, M. P., Havel, T. F. & Wüthrich, K. Solution conformation of proteinase inhibitor IIA from bull seminal plasma by 1H nuclear magnetic resonance and distance geometry. J Mol Biol 182, 295–315 (1985).
3. Li, F., Lee, J. H., Grishaev, A., Ying, J. & Bax, A. High accuracy of Karplus equations for relating three-bond J couplings to protein backbone torsion angles. Chemphyschem 16, 572–578 (2015).
4. Havel, T. & Wüthrich, K. A distance geometry program for determining the structures of small proteins and other macromolecules from nuclear magnetic resonance measurements of intramolecular1H−1H proximities in solution. Bltn Mathcal Biology 46, 673–698 (1984).
5. Rule, G. & Hitchens, K. Fundamentals of Protein NMR Spectroscopy. vol. 5 (Springer-Verlag, 2006).
6. Havel, T. F. & Wüthrich, K. An evaluation of the combined use of nuclear magnetic resonance and distance geometry for the determination of protein conformations in solution. J Mol Biol 182, 281–294 (1985).