- О компании
- Направления деятельности
-
Медиатека
СтатьиСервис

10.06.2025
Сервисная сторона лаборатории: от инсталляции оборудования до постгарантийной поддержки
Интервью со специалистами технической поддержки Компании Хеликон.
СтатьиИнтервью
27.03.2024
Интервью Анатолия Смирнова телеканалу PROБизнес
«В любой биологической лаборатории в России есть что-то от нас».
- Новости
- Мероприятия
- Партнеры




.jpg "Реализованные проекты")
Медиатека
От генома к популяции: как геномика может помочь сохранить биоразнообразие
Оглавление:
•
Геномные методы в исследованиях биоразнообразия
•
Бардкодирование и метабаркодирование ДНК
•
Секвенирование ограниченного набора геномных локусов
•
Данные экспрессии генов
•
Полногеномное секвенирование
•
Неинвазивный отбор геномов
Сегодня Земля находится на пороге шестого в истории планеты массового вымирания видов, которое будет иметь разрушительные последствия для функционирования, стабильности и эволюции природных систем и будущего человечества 1. Мероприятия по сохранению биоразнообразия требуют постоянного мониторинга состояния популяций и экосистем, понимания генетического разнообразия, адаптивного потенциала и взаимодействия особей различных таксономических групп, а также контроля влияния деятельности человека на среду обитания и виды, которые находятся под угрозой исчезновения. Всё это становится возможным благодаря кооперации учёных из разных областей – эволюционной биологии, зоологии и ботаники, экологии и молекулярной экологии, генетики и геномики.
Такое междисциплинарное взаимодействие позволяет наблюдать за состоянием экосистем под различным углом. На «макроуровне» полевые биологи отслеживают глобальную динамику популяций – наблюдают за изменениями в поведении животных в естественной среде обитания, получают биологические образцы и ведут учёт существующих, новых и исчезающих видов. Молекулярные экологи и генетики получают данные, которые недоступны невооружённому глазу, – на «микроуровне».
И хотя именно генетические различия лежат в основе биологического разнообразия, только в последние годы геномика начала выходить за рамки отдельных исследований и изучения модельных организмов в лабораторных условиях и находить практическое применение для решения «глобальных» проблем. Сегодня данные генетики и геномики активно используются для создания программ, направленных на сохранение видов и популяций, включая разработку стандартизированных протоколов учёта генетического разнообразия и адаптивного потенциала организмов, а также планирование конкретных шагов к достижению целей 2.
Геномные методы в исследованиях биоразнообразия
В арсенале учёных имеется большой набор инструментов, позволяющих детально исследовать генетические различия организмов разных таксономических групп. При этом каждый подход предназначен для конкретной области применения. Одним из наиболее распространённых методов генетики и геномики остаётся секвенирование. Данный метод стремительно развивается и совершенствуется – современные технологии высокопроизводительного секвенирования позволяют считывать фрагменты ДНК до >10 000 пар нуклеотидов.
Для изучения биоразнообразия всё большую популярность набирают метабаркодирование ДНК (ДНК-штрихкодирование), секвенирование транскриптома (RNA-Seq) и полногеномное секвенирование (Рис. 1). Эффективность подходов зависит от множества факторов: количества и качества доступного биологического материала, оснащённости лабораторий, владения методами биоинформатики для обработки данных, наличия и полноты данных эталонных геномов, а также затрат на исследования 3.
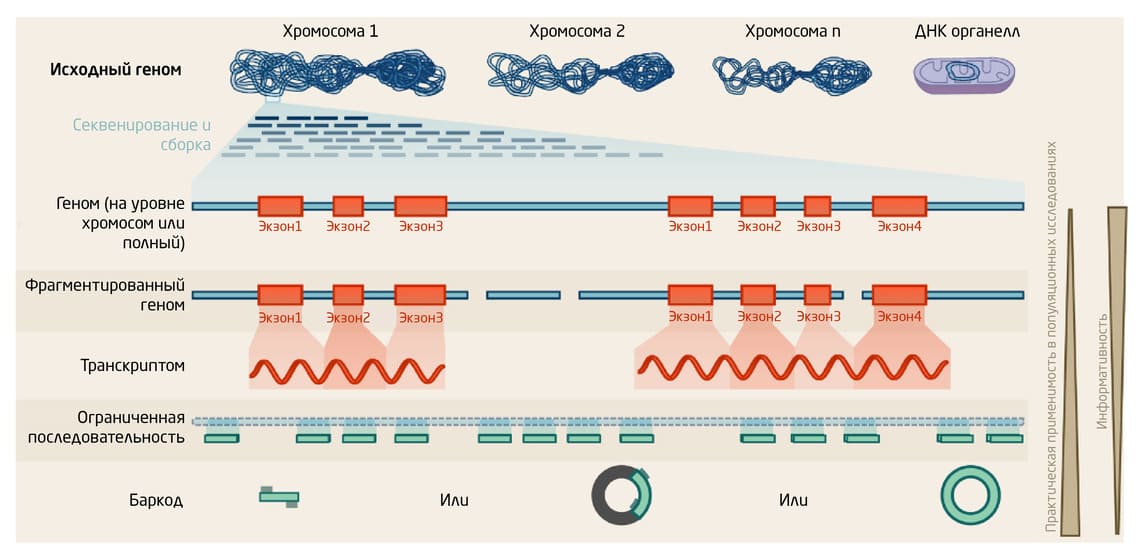
Рис. 1. Принципы основных методов геномики, используемых в исследованиях биоразнообразия
3.
Бардкодирование и метабаркодирование ДНК
ДНК-штрихкодирование используется для определения видовой принадлежности организмов и мониторинга биологического разнообразия. Основная концепция штрих-кодирования ДНК довольно проста: образец неизвестной таксономической группы может быть идентифицирован путём предварительного секвенирования и последующего сравнения определённой области ДНК (штрих-кода) с референсным геномом, который содержит те же области ДНК, что и исследуемый образец 4.
ДНК-баркодирование включает три основных этапа. Первый и ключевой этап – выделение ДНК. От качества выделенной молекулы будет зависеть точность и эффективность последующих шагов. С использованием универсальных праймеров проводится ПЦР-амплификация и секвенирование участков определённого гена. Полученная последовательность интересующего участка гена (400-800 п.н.) и будет баркодом данного организма 5, 6. При этом баркоды должны удовлетворять ряду условий. Они должны содержать информативные локусы ДНК и иметь «универсальные», или таксон-специфичные, праймеры, комплементарные консервативным областям различных видов, но в то же время обладать высокой изменчивостью (не по длине, чтобы избежать трудностей с выравниванием последовательности и её идентификацией) при относительно невысокой скорости замен внутри видов. Это позволит различать близкородственные виды 7.
Например, для баркодирования беспозвоночных используется ген субъединицы I митохондриальной цитохромоксидазы С (COI). Для штрих-кодирования геномов позвоночных могут применяться гены рибосомных субъединиц 12S и 16S и ген цитохрома b митохондриальной ДНК; для баркодирования растений – гены rbcL и matK ДНК хлоропластов; для баркодирования грибов – последовательности внутренних транскрибируемых спейсеров ITS рибосомной ДНК; для простейших и нематод – ген субъединицы 18S рибосомной ДНК (рДНК), а для бактерий – ген субъединицы 16S рДНК 8.
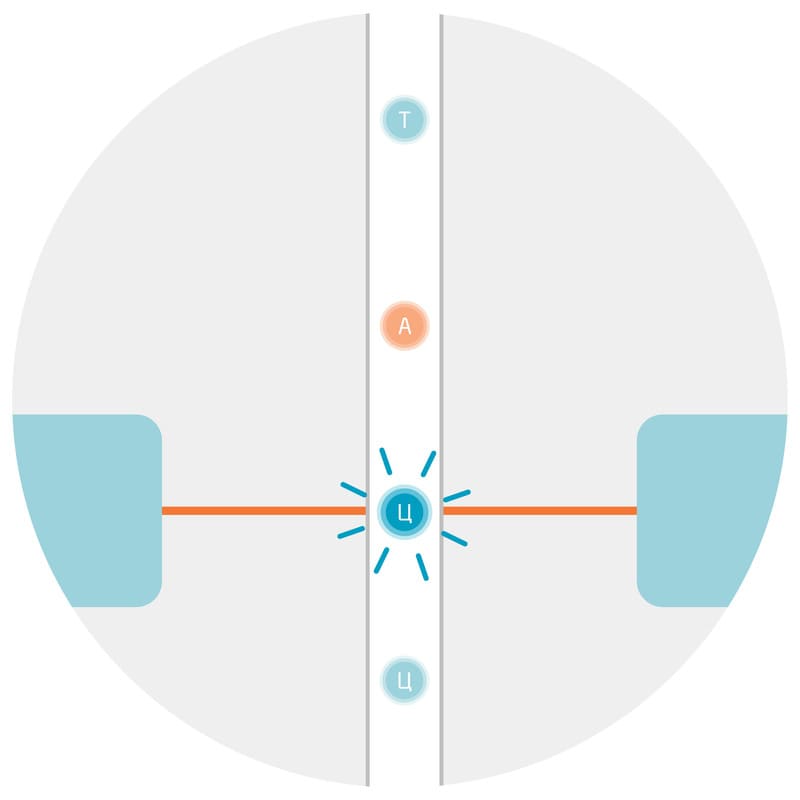
Как и гены, праймеры ПЦР специфичны для конкретной таксономической группы, а значит таксономическую принадлежность (рыбы, птицы, млекопитающие и т.д.) интересующего организма необходимо определять до проведения анализа. Например, по морфологическим признакам. Если геном организма был отсеквенирован ранее, последовательность баркода можно определить из полной последовательности генома (Рис. 2).
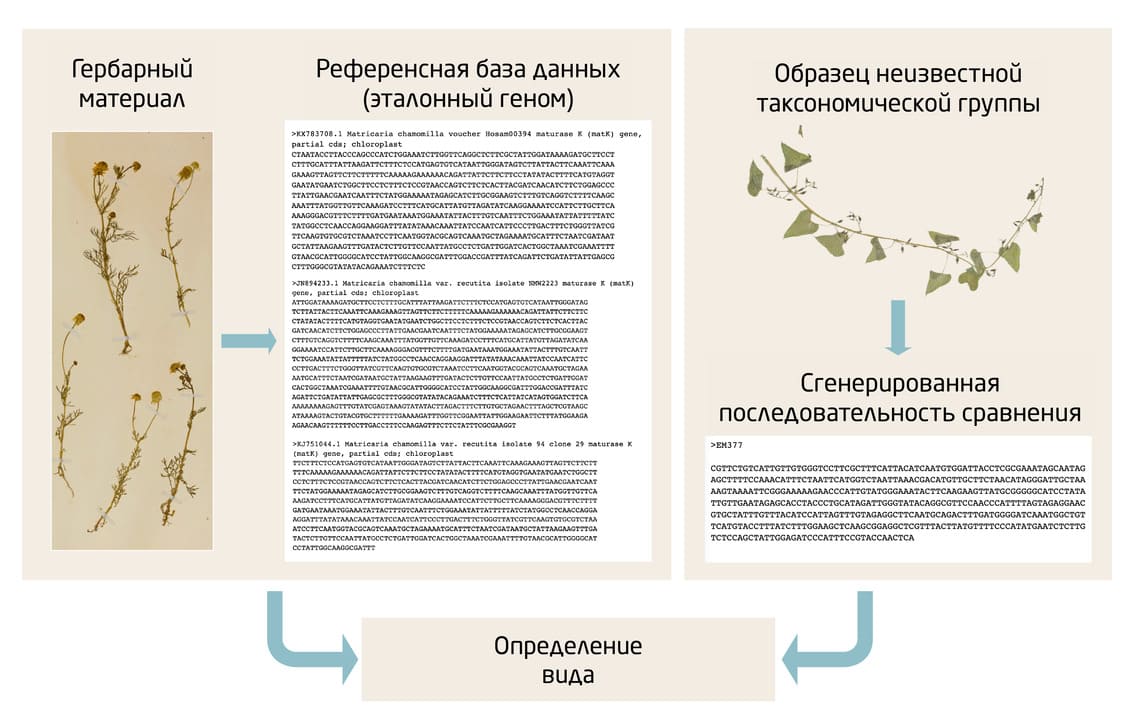
Рис. 2. Упрощённая схема принципа баркодирования на примере определения видовой принадлежности лекарственных растений
4.
После амплификации образцы отправляются на секвенирование. С помощью последующего биоинформатического анализа ДНК-баркоды исследуемых организмов сравниваются с существующей базой данных (Рис. 3).
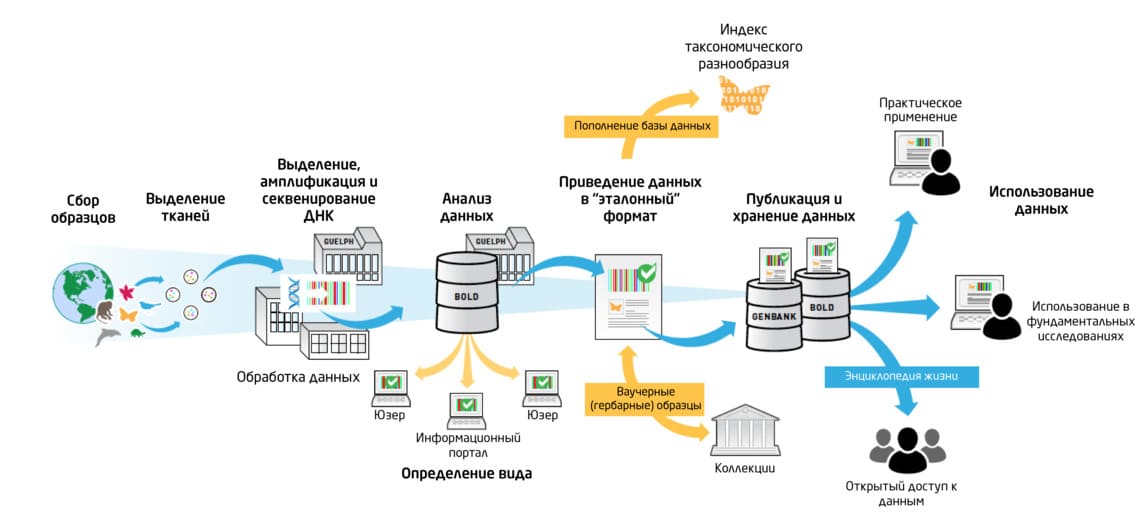
Рис. 3. Основные этапы баркодирования – от получения образцов до практического применения данных
5.
С помощью ДНК-баркодирования можно проводить оценку видового разнообразия в масштабах целых экосистем практически во всех средах обитания. Однако метод не лишён недостатков. Короткая длина областей ДНК, используемых для штрих-кодирования, часто не позволяет получить точную характеристику генетического и, как следствие, таксономического разнообразия организмов внутри изучаемого сообщества, разграничить близкородственные виды и виды, содержащие интрогрессированные генетические последовательности или органелларные гены.
Одно из решений – скимминг генома. Это метод секвенирования нового поколения, позволяющий считывать высококопийную фракцию генома и получать нуклеотидные последовательности геномов хлоропластов, фрагментов митохондриальной ДНК, а также ядерной рДНК 9. Скимминг может применяться для изучения повреждённых образцов ДНК (например, у музейных экспонатов), которые сложно анализировать с помощью «классических» методов секвенирования.
Метабаркодирование ДНК представляет собой комбинацию высокопроизводительного секвенирования и ДНК-баркодирования. Метод позволяет идентифицировать одновременно несколько таксонов с использованием внеклеточной и (или) тотальной ДНК, извлечённой из образцов, содержащих ДНК различного происхождения 4.
Секвенирование ограниченного набора геномных локусов
Методы секвенирования ДНК с использованием ограниченного набора геномных локусов (Reduced representation DNA sequencing, RRS), или «генотипирование путём секвенирования», широко применяются в исследованиях немодельных видов 10. И хотя они охватывают лишь небольшую часть генома, полученных данных, как правило, достаточно для оценки генетического разнообразия, инбридинга, размера и структуры популяции, потока генов внутри и между популяциями, выявления филогенетических взаимоотношений между видами и анализа филогеографических паттернов.
Метод заключается в уменьшении количества геномных данных, используемых для секвенирования, с помощью рестрикционных эндонуклеаз, отбора фрагментов ДНК по размеру или же гибридизации с использованием специальных зондов (например, ультраконсервативных элементов) и последующего секвенирования полученных участков ДНК. Несмотря на преимущества метода – относительную простоту и невысокую стоимость – сопоставление данных из разных исследований требует идентичности экспериментальных протоколов, что значительно ограничивает воспроизводимость результатов. Также при использовании эндонуклеаз рестрикции важно учитывать возможность «потери» аллелей. Для улучшения интерпретации данных и идентификации локусов, содержащих однонуклеотидные полиморфизмы, рекомендуется сопоставлять их с эталонными геномами 3.
Данные экспрессии генов
Данные об экспрессии генов, полученные, например, с помощью секвенирования РНК (RNA-Seq), используются для оценки генетической и функциональной изменчивости, включая адаптацию организмов к меняющимся условиям среды на уровне отдельных особей и популяций, а также помогают выявлять гены-кандидаты, играющие «движущую» роль в экологических и эволюционных процессах. Анализ транскриптома позволяет понимать реакцию животных на воздействие токсинов (например, родентицидов) 11 и восприимчивость или устойчивость к вирусным инфекциям 12, прогнозировать сдвиги ареалов обитания, а также выявлять уязвимые популяции или, наоборот, особей, которые успешно адаптируются к меняющимся условиям среды.
Учитывая, что РНК легко подвергается деградации и разрушению, а сам процесс транскрипции зависит множества факторов, включая тип клеток, возраст и пол организма, физиологическое состояние и даже циркадные ритмы, транскриптомный анализ – более сложная задача, чем секвенирование ДНК. Вместе с секвенированием РНК часто применяются исследования эпигенома, ведь именно эпигенетические модификации контролируют фенотипическую изменчивость и играют ключевую роль в быстрых адаптивных реакциях в ответ на глобальные экологические изменения. Именно поэтому их изучение чрезвычайно важно для выявления видов, уязвимых к изменениям окружающей среды, а также мониторинга эволюции инвазивных видов 3.
Полногеномное секвенирование
Благодаря высокой скорости и точности результатов полногеномное секвенирование (Whole Genome Sequencing, WGS), пожалуй, один из самых эффективных и широко используемых методов молекулярной генетики и геномики. Современные секвенаторы позволяют получить 48 Гб геномных данных из небольших геномов всего за 12 часов и до 6 Тб данных за 24 часа в случае больших геномов с точностью 99.9%!
С помощью данных WGS можно решать следующие задачи:
- проводить анализ селекционного материала;
- обнаруживать мутации в регуляторных элементах, редкие варианты и структурные вариации генов;
- выявлять генетическую основу фенотипических признаков (например, восприимчивость или устойчивость исчезающих видов к болезням) и видовой диверсификации;
- изучать механизмы естественного отбора и историю эволюции вымерших видов, а также внутривидовое генетическое разнообразие и структуру предковых форм.
Всё это крайне важно для сохранения и восстановления биоразнообразия.
Кроме того, высокая мощность и разрешение метода позволяют амплифицировать геномную информацию из образцов вымерших видов или видов, находящихся под угрозой исчезновения. При этом важно помнить, что анализ повреждённых и фрагментированных образцов ДНК музейных или ископаемых экспонатов, которые в большинстве случаев не превышают размера в 100 п.н., требует наличия эталонных геномов для картирования ридов, полученных в процессе полногеномного секвенирования 13.
Наиболее эффективным с экономической точки зрения методом WGS в популяционных геномных исследованиях на сегодняшний день является Pool-Seq, основанный на смешивании ДНК нескольких организмов и глубоком секвенировании объединённых геномов (Рис. 4) 14. Pool-Seq популярен при изучении эволюции микроскопических организмов с небольшим (до 1 Гб) геномом и для определения частоты аллелей редких генетических вариантов. Особенно эффективен этот подход при работе сразу с несколькими популяциями, в каждой из которых отбирается большое количество особей (> 50) для оценки генетического разнообразия, адаптации, географической дифференциации популяций и взаимосвязей между изменениями в генотипе и окружающей среде 3. Несмотря на преимущества и экономическую эффективность Pool-Seq, надёжность и точность получаемых результатов напрямую зависят от размера пула и снижаются в сложных системах со слабовыраженной популяционной структурой 15.
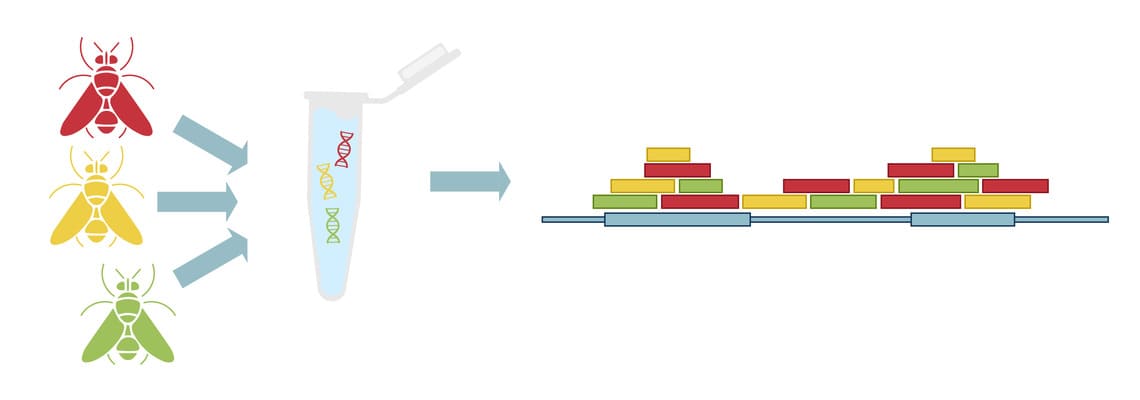
Рис. 4. Схематическое представление метода Pool-Seq.
Неинвазивный отбор геномов
Неинвазивный или минимально инвазивный отбор (сэмплинг) заключается в анализе проб, полученных из биологического материала, например, из фекалий, перьев или волос 16. Несмотря на широкое применение метода для мониторинга состояния экосистем и получения информации об экологии исчезающих видов, его недостатки очевидны – неинвазивно отобранные образцы в большинстве случаев содержат небольшое количество некачественной ДНК, к тому же часто загрязнённой экзогенной ДНК. Некоторые проблемы удаётся решать с помощью дополнительных подходов, позволяющих увеличивать количество извлекаемой эндогенной, или «целевой», ДНК. К таким подходам относятся:
- генотипирование с использованием массивов однонуклеотидных полиморфизмов (SNP), при котором в качестве зондов используются последовательности ДНК в областях известных SNP 17;
- секвенирование ампликонов, полученных после мультиплексной ПЦР (этот тип ПЦР предполагает одновременную амплификацию в одной пробирке сразу нескольких последовательностей ДНК), которое позволяет одновременно анализировать большое количество SNP-маркеров 18;
- таргетное секвенирование, или захват целевой последовательности генов, с использованием олигонуклеотидов РНК или ДНК, определённых заранее с помощью метода RRS или из эталонных геномов 19.
С каждым годом антропогенное воздействие на природу усиливается, а изменения окружающей среды происходят всё быстрее. Современные методы геномики и накопленные геномные данные обеспечивают основу для разработки мер по сохранению биоразнообразия, что невозможно без учёта и знания генетического разнообразия видов. С практической точки зрения эти данные помогают разрабатывать стратегии по улучшению адаптивного потенциала отдельных особей и жизнеспособности популяций. При этом важнейшим компонентом большинства методов геномики остаётся доступность эталонных геномов, полученных из высококачественных образцов ДНК. Именно они формируют основу для количественной оценки биоразнообразия, расширяют возможности отдельных методов и интерпретацию полученных геномных данных.

Литература
1. Cowie R.H. et al. The Sixth Mass Extinction: fact, fiction or speculation? Biological Reviews of the Cambridge Philosophical Society, 2022.
2. Nielsen E.S. et al. Molecular ecology meets systematic conservation planning. Trends in Ecology & Evolution, 2023.
3. Theissinger K. et al. How genomics can help biodiversity conservation. Trends in Genetics, 2023.
4. Raclariu-Manolică A.C., de Boer H.J. Evidence-Based Validation of Herbal Medicine (Second Edition). Chapter 8 – DNA barcoding and metabarcoding for quality control of botanicals and derived herbal products. Translational Research on Botanicals, 2022.
5. Carmon J.L. et al. Identification of Unknown Organisms by DNA Barcoding: A Molecular Method for Species Classification. Research and Development, Office Invasive Mussels, Final Report, 2014.
6. Cyanobacteria. From Basic Science to Applications. Edited by Mishra A.K., Tiwari D.N. and Rai A.N., Academic Press, 2019.
7. Шеховцов С.В. и др. ДНК-штрихкодирование: методы и подходы. Успехи современной биологии, 2019.
8. Kress W.J. et al. DNA barcodes for ecology, evolution, and conservation. Trends in Ecology & Evolution, 2015.
9. Straub S.C.K. et al. Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. American Journal of Botany, 2012.
10. Hohenlohe P.A. et al. Population genomics for wildlife conservation and management. Molecular Ecology, 2021.
11. Fraser D. et al. Genome-wide expression reveals multiple systemic effects associated with detection of anticoagulant poisons in bobcats (Lynx rufus). Molecular Ecology, 2018.
12. Campbell L.J. et al. A novel approach to wildlife transcriptomics provides evidence of disease-mediated differential expression and changes to the microbiome of amphibian populations. Molecular Ecology, 2018.
13. de Manuel M. et al. The evolutionary history of extinct and living lions. Proceedings of the National Academy of Sciences of the United States of America, 2020.
14. Anand S. et al. Next Generation Sequencing of Pooled Samples: Guideline for Variants' Filtering. Scientific Reports, 2016.
15. Dorant Y. et al. Comparing Pool-seq, Rapture, and GBS genotyping for inferring weak population structure: The American lobster (Homarus americanus) as a case study. Ecology and Evolution, 2019
16. Ruiz-Lopez M.J. et al. A novel landscape genetic approach demonstrates the effects of human disturbance on the Udzungwa red colobus monkey (Procolobus gordonorum). Heredity, 2016.
17. LaFramboise T. Single nucleotide polymorphism arrays: a decade of biological, computational and technological advances. Nucleic Acids Research, 2009.
18. Onda Y. et al. Multiplex PCR Targeted Amplicon Sequencing (MTA-Seq): Simple, Flexible, and Versatile SNP Genotyping by Highly Multiplexed PCR Amplicon Sequencing. Frontiers in Plant Science, 2018.
19. Ribeiro G.P. et al. A bioinformatic platform to integrate target capture and whole genome sequences of various read depths for phylogenomics. Molecular Ecology, 2021.