



.jpg "Реализованные проекты")


Медиатека
Секвенирование микроРНК
Оглавление:
•
Почему стоит выбрать именно этот метод анализа?
•
Как обеспечить надёжность NGS-данных для образцов из биожидкостей?
•
Подготовка NGS-библиотек
1. Очистка с помощью магнитных частиц вместо вырезания из геля
2. Как избежать погрешностей секвенирования с помощью УМИ
•
Покрытие секвенирования для микроРНК из биожидкостей
•
Установка порогового значения TPM
•
Нормализация для анализа дифференциальной экспрессии
•
IPA для интерпретации сложных «омных» данных
2. Как избежать погрешностей секвенирования с помощью УМИ
Почему стоит выбрать именно этот метод анализа?
Важный выбор, стоящий перед исследователем, – это выбор платформы для анализа. Для исследований методом микрочипов необходимо такое исходное количество РНК, какое невозможно получить из образцов биожидкостей, а чувствительность и динамический диапазон метода недостаточны.
Количественная ПЦР – популярный аналитический инструмент с высокой чувствительностью и специфичностью, но мишени исследования должны быть известны заранее. Секвенирование нового поколения (NGS, next generation sequencing) не требует предварительной информации об отдельных микроРНК, присутствующих в образцах, и охватывает все последовательности нужного размера и характеристик. Это экономически эффективный подход к профилированию всего массива микроРНК, благодаря которому удаётся обнаруживать новые микроРНК, а также малые РНК других типов, являющиеся потенциальными биомаркерами, – например, пиРНК (piRNA, piwi-interacting RNA), – что делает NGS предпочтительной технологией для скрининга. Новые методы подготовки NGS-библиотек микроРНК, использующие уникальные молекулярные индексы или идентификаторы (УМИ), позволяют преодолеть погрешность в количественной оценке.
Как обеспечить надёжность NGS-данных для образцов из биожидкостей?
1. Контрольные шпильки для NGS
Проведение достаточно чувствительного и точного NGS-анализа микроРНК из биожидкостей – сложная задача, для решения которой важно использовать протоколы выделения РНК и контроля качества, оптимизированные для образцов такого типа (детальное описание протоколов представлено в соответствующих разделах). Добавление синтетической шпилечной микроРНК во время выделения РНК повышает выход и воспроизводимость результатов
1. Однако количество шпилечной микроРНК следует оптимизировать, чтобы не тратить на неё слишком много прочтений при секвенировании. Для облегчения анализа данных шпилечная РНК должна относиться к виду, достаточно далеко отстоящему от исследуемого вида в эволюционном смысле.
Библиотека контрольных шпилек QIAseq miRNA Library – набор из 52 последовательностей микроРНК резуховидки Таля (Arabidopsis thaliana) – отвечает этим требованиям при использовании NGS-анализа образцов микроРНК человека и других млекопитающих. Успешность и однородность экстракции можно оценить в ходе количественной ПЦР, специфичной для шпилек. На этапе обратной транскрипции с помощью шпилек можно выявить любой ингибитор ферментов, перенесённый из образца. Это позволяет предвидеть, какие факторы повлияют на создание библиотеки и последующее секвенирование, и подтвердить любой негативный эффект, проверив данные секвенирования для 52 шпилек (Рис. 1).
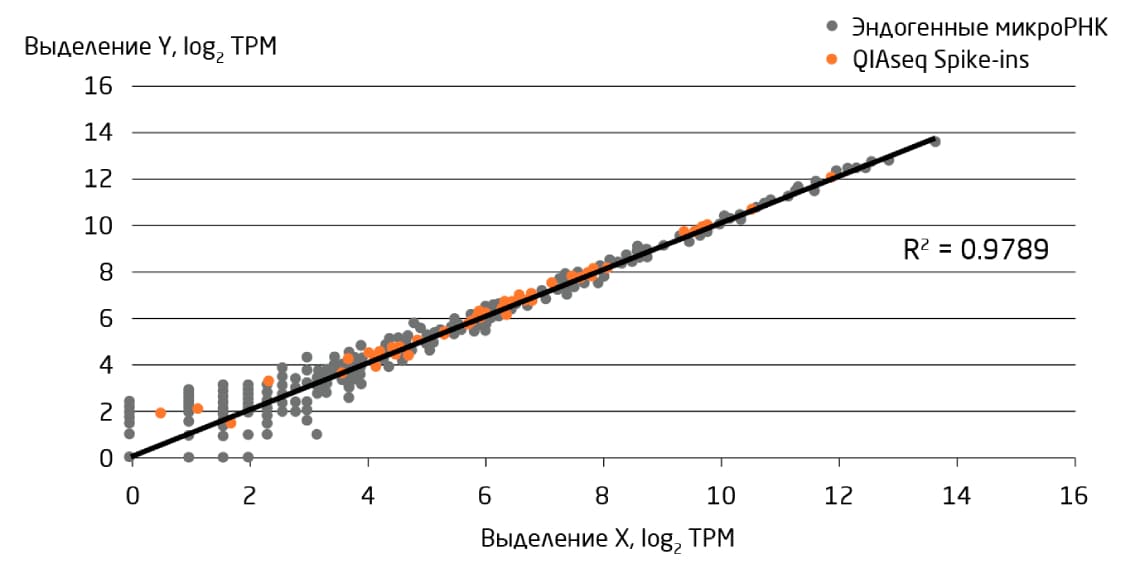
Рис. 1. Воспроизводимость и оценка линейности распределения микроРНК и малых РНК из биожидкости для рабочего процесса NGS – от выделения РНК до секвенирования. РНК выделили в двух независимых экспериментах с использованием одного и того же пула образцов сыворотки/плазмы, после чего подготовили две независимых библиотеки и провели секвенирование. Этот график демонстрирует превосходную воспроизводимость, о чем свидетельствует корреляция между 52 РНК-шпильками, добавленными во время процедуры выделения РНК. ММКП – метки на миллион картированных прочтений.
Подготовка NGS-библиотек
1. Очистка с помощью магнитных частиц вместо вырезания из геля
До сих пор секвенирование микроРНК было сопряжено со многими трудностями. Для подготовки библиотек нередко требовалась утомительная очистка геля для удаления адаптер-димеров и загрязняющей РНК. Электрофорез с последующим вырезанием нужной полосы из геля занимает много времени и добавляет, как минимум, один день к и без того продолжительным экспериментам. Кроме того, вырезание полос может сильно варьировать от образца к образцу и от пробы к пробе. Такая вариабельность отрицательно сказывается на качестве данных. Для большей воспроизводимости и ускорения процесса мы рекомендуем проводить очистку с помощью магнитных частиц с помощью набора QIAseq miRNA Library Kit, который содержит запатентованные реагенты для удаления адаптер-димеров и загрязняющих РНК.

Таблица 1. Сравнение эффективности и общей производительности секвенирования, достигнутых с помощью метода QIAGEN и методов трех других ведущих поставщиков наборов для подготовки библиотек. Чтобы оценить эффективность секвенирования, для каждой из методик был рассчитан процент прочтений, прошедших фильтр при усечении адаптера и картировании. Зеленые галочки указывают на удовлетворительную, желтые – на среднюю эффективность в соответствующей категории. Красные крестики указывают либо на результат ниже среднего, либо, в случае ингибирования YRNA, на недоступность опции. *Требуется разделение в геле 2.
Стандартная процедура (Рис. 2) не требует очистки в геле, вырезания полос и элюирования, что сокращает время работы и упрощает рабочий процесс. Кроме того, технология, используемая в наборе QIAseq miRNA Library Kit, блокирует включение в библиотеку секвенирования продуктов малых РНК человека Y4, в изобилии присутствующих в сыворотке и плазме крови 3, тем самым снижая процент напрасно потраченных прочтений.

Рис. 2. Рабочий процесс подготовки библиотеки для РНК-секвенирования с помощью набора QIAseq miRNA Library Kit. Усовершенствованная стратегия подготовки библиотек использует УМИ для отображения исходного количества молекул РНК и устранения погрешности ПЦР. Очистка с использованием магнитных частиц позволяет автоматизировать рабочий процесс на роботизированной платформе.
2. Как избежать погрешностей секвенирования с помощью УМИ
Существенный вклад в погрешность секвенирования в процессе создания библиотеки малых РНК вносит последующая ПЦР, поскольку эффективность амплификации может быть разной для разных матриц. Протокол подготовки библиотеки с помощью набора QIAseq miRNA Library Kit позволяет избежать погрешности путем введения УМИ. Включение УМИ в последовательности происходит при подготовке библиотеки с помощью праймеров для обратной транскрипции, несущих УМИ длиной 12 нуклеотидов. Поскольку в каждой позиции может стоять один из четырёх нуклеотидов, существует около 17 миллионов различных 12-нуклеотидных комбинаций, то есть любая отдельная РНК может получить собственную метку УМИ (Рис. 2).
Секвенирование традиционных библиотек микроРНК даёт сырой массив прочтений, количество которых часто обусловлено погрешностью ПЦР, а не количеством копий исходной РНК. Это явление проиллюстрировано рисунком 3, где результаты показывают 6 прочтений для образца 1 и 3 прочтения для образца 2, или соотношение 6:3 (2:1) вместо истинного соотношения транскриптов в образце (3:1).
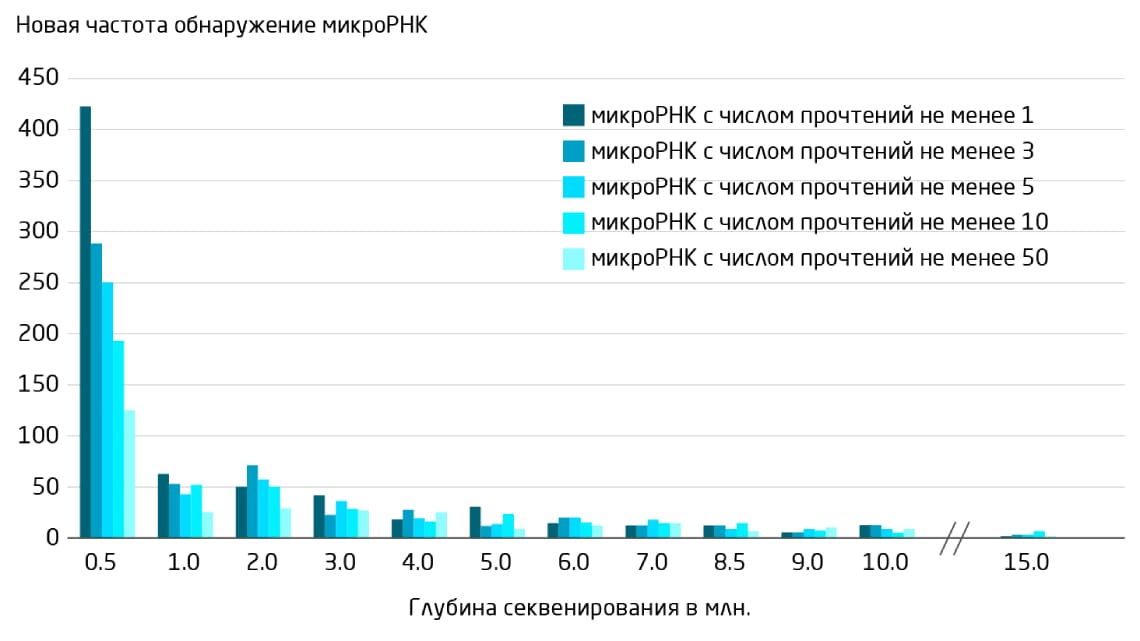
Рис. 3. Коррекция погрешности ПЦР с помощью УМИ. A. Без УМИ точное соответствие между количеством прочтений и количеством копий исходной РНК отсутствует. B. С УМИ данные точно отражают количество исходного транскрипта.
Количественная оценка прочтений на основе УМИ устраняет погрешность, позволяя данным отражать исходное количество захваченных молекул РНК. Если рассматривать все копии одного УМИ как одну исходную РНК, данные секвенирования можно привести к первоначальным соотношениям. Использование УМИ позволяет точнее интерпретировать количество генов и изменения в их экспрессии.
Покрытие секвенирования для микроРНК из биожидкостей
Количество прочтений, измеренное с помощью NGS для определенной последовательности микроРНК, не связано напрямую с ее количеством
4, поэтому NGS не подходит для абсолютной количественной оценки. Для выявления различий между образцами по относительной экспрессии данные NGS, как и данные анализа микрочипов и количественной ПЦР, необходимо нормализовать и проанализировать. Кроме того, следует учитывать, что измерение конкретной микроРНК с помощью NGS не происходит независимо от других микроРНК. Если какая-то определенная микроРНК присутствует в большом количестве, это может уменьшить количество прочтений для обнаружения других микроРНК. Необходимо заботиться о том, чтобы покрытие секвенирования было достаточным для точного анализа микроРНК, экспрессированных на низком уровне, какие обычно встречающихся в биологических жидкостях.
Покрытие прочтений для библиотеки микроРНК – один из наиболее важных факторов и для анализа дифференциальной экспрессии, и для выявления новых микроРНК. На Рис. 4 представлены результаты анализа данных NGS для микроРНК из ткани икроножной мышцы мышей и из соответствующих образцов плазмы с использованием различного количества сырых некартированных прочтений (от 0,5 до 15 миллионов)
5. Секвенирование с большим покрытием позволяет идентифицировать новые предполагаемые микроРНК. По опыту QIAGEN идеальное число для охвата микроРНК из биожидкостей – 12 миллионов прочтений.
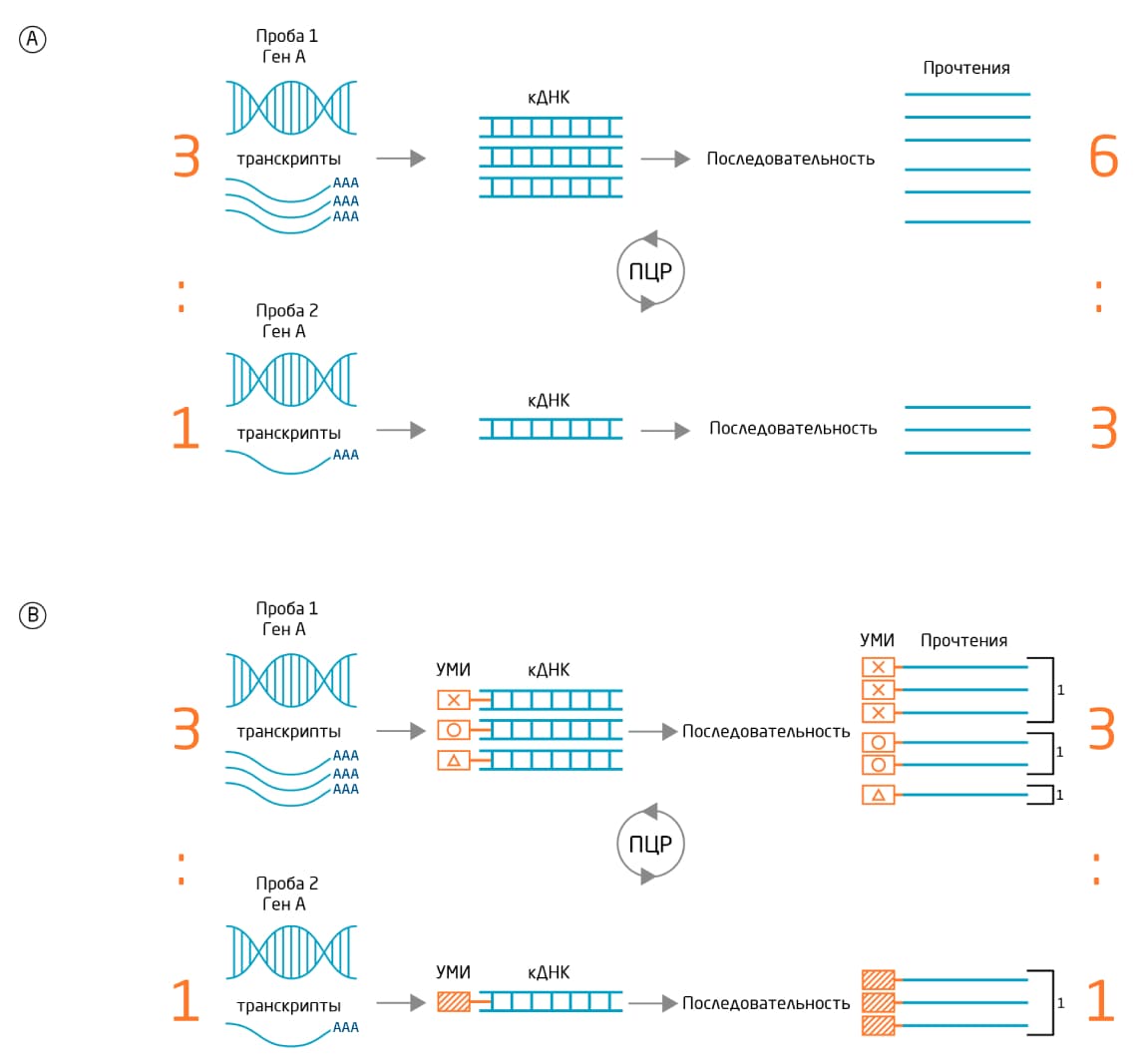
Рис. 4. Влияние покрытия секвенирования на частоту обнаружения редких или новых микроРНК. По мере добавления к покрытию секвенирования по 1 миллиону прочтений (отфильтрованных, но не картированных) количество дополнительно обнаруженных новых микроРНК уменьшается. Каждый столбец на диаграмме показывает количество новых обнаруженных микроРНК, имеющих, по крайней мере, 1, 3, 5, 10 или 50 прочтений, соответственно, при различном покрытии секвенирования 5.
Установка порогового значения TPM
В идеале количество прочтений микроРНК в разных образцах одного исследования должно быть одинаковым, чтобы сравнивать образцы друг с другом. Используемая процедура нормализации должна учитывать любые различия в количестве прочтений между образцами, независимо от покрытия секвенирования (Рис. 5). Расчёт количества меток на миллион сопоставленных прочтений (TPM, tags per million) является средством нормализации к общему количеству сопоставленных прочтений. Прочтения, обнаруженные при TPM ˂1, могут представлять собой микроРНК, экспрессированные на низком уровне, либо артефакты, в то время как прочтения, обнаруженные при TPM ˂5, бывает трудно подтвердить другими методами (например, количественной ПЦР), и их выявление обычно нельзя улучшить путем увеличения покрытия секвенирования. Установление порога TPM – полезный способ сосредоточиться на наиболее надежных прочтениях микроРНК.
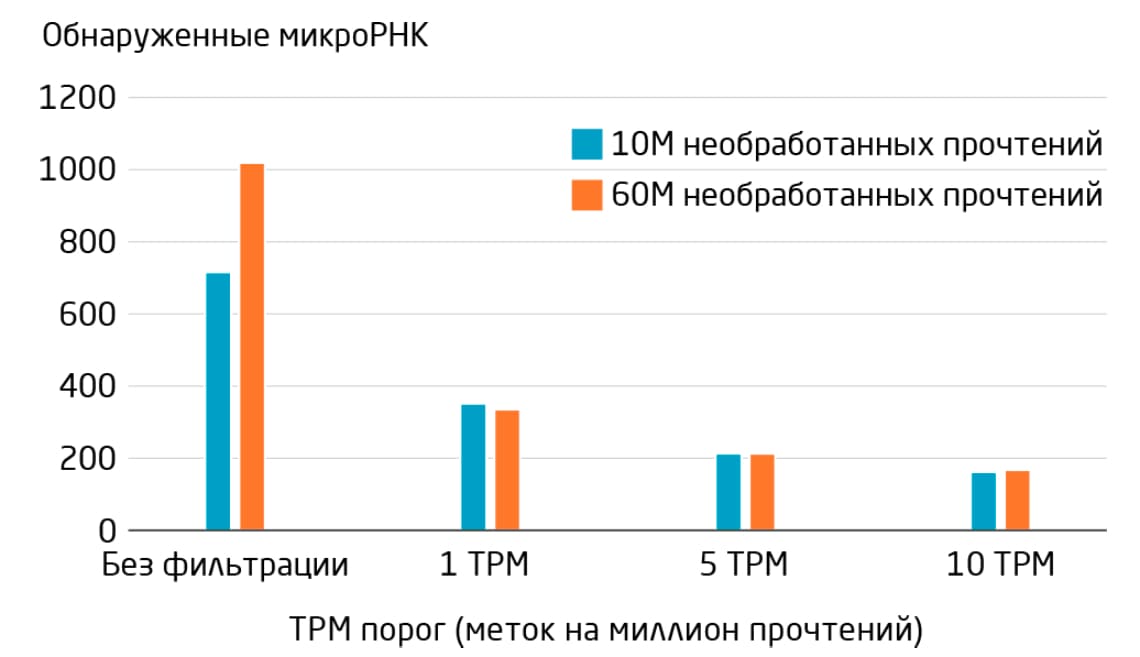 <
<
Рис. 5. Вычисление TPM сопоставленных прочтений для коррекции нормализации при различном покрытии секвенирования. Объединенные сыворотка и плазма были секвенированы с 10М или 60М необработанных прочтений на образец. После нормализации количество обнаруженных микроРНК одинаково. Применение порогового значения позволяет сфокусироваться на наиболее надежных сигналах.
Нормализация для анализа дифференциальной экспрессии
Нормированные значения TPM позволяют оценить количество РНК в образце, однако сравнение между собой отдельных образцов или их групп требует другого подхода к нормализации – такого, который компенсирует различия в покрытии секвенирования, а также эффекты избыточной и недостаточной выборки. Существует множество методов нормализации данных секвенирования РНК для анализа дифференциальной экспрессии генов. Эти подходы часто предполагают, что большинство генов одинаково экспрессируются в разных группах образцов. В исследовании, посвящённом сравнению методов нормализации
6, ни один из результатов, полученных с помощью этих методов, существенно не отличался от результатов нормализации с использованием усечённого среднего значения M (TMM, trimmed mean of M values).
TMM позволяет рассчитать эффективные размеры библиотек, используемые затем как часть нормализации для выборки. TMM-нормализация регулирует размеры библиотек, исходя из предположения, что большинство генов не подвержены дифференциальной экспрессии. Коэффициенты ТММ используются для получения нормализованных количеств прочтений
7. TMM-нормализация использована в потоковых анализах CLC Genomics Workbench miRNA analysis (Рис. 6), являющихся частью плагина Biomedical Genomics Analysis. Результаты анализа дифференциальной экспрессии визуализируются в виде диаграммы Венна и тепловых карт. Из CLC Genomics Workbench сравнение экспрессии исходных микроРНК можно загрузить непосредственно в Ingenuity® Pathway Analysis (IPA®).

Рис. 6. Схема полного цикла обработки данных NGS для микроРНК, доступного через CLC Genomics Workbench при использовании плагина Biomedical Genomics Analysis.
Оптимизированный рабочий процесс и описанные протоколы – неотъемлемая часть услуг QIAGEN по NGS для микроРНК биожидкостей. Этот рабочий процесс дает оптимальные результаты и является наилучшим подходом для работы с микроРНК. В частности, с микроРНК биожидкостей.
IPA для интерпретации сложных «омных» данных
Биологическая интерпретация высококачественных достоверных данных, полученных в «омных» исследованиях, требует использования передовых методов биоинформатики, таких как Ingenuity Pathway Analysis от компании QIAGEN. Программное обеспечение IPA – мощный аналитический и поисковый инструмент, раскрывающий значение таких данных и позволяющий выявить в биологических системах новые мишени и потенциальные биомаркеры 8. IPA широко используется в области наук о жизни и упоминается в тысячах публикаций.

Литература
1. Andreasen D. et al. Improved microRNA quantification in total RNA from clinical samples. Methods, 2010.
2. Coenen-Stass A.M.L. et al. Evaluation of methodologies for microRNA biomarker detection by next generation sequencing. RNA Biology, 2018.
3. Dhahbi J.M. Circulating small noncoding RNAs as biomarkers of aging. Ageing Research Reviews, 2014.
4. Hafner M. et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA, 2011.
5. Metpally R.P.R. et al. Comparison of analysis tools for miRNA high throughput sequencing using nerve crush as a model. Frontiers in Genetics, 2013.
6. Seyednasrollah F., Laiho A., Elo L.L. Comparison of software packages for detecting differential expression in RNA-seq studies. Briefings in Bioinformatics, 2015.
7. Dillies,M.A. et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Briefings in Bioinformatics, 2013.
8. Krämer A. et al. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics, 2014.























